自闭症谱系障碍(ASD)是一种“广泛性发育障碍”,其特征是“沟通和社会交往异常”和“受限制的重复性活动和兴趣”。这种疾病的发展从婴儿期就很明显,大多数病例在5岁时就被发现。2010年对17185名被诊断为自闭症谱系障碍的儿童进行的分析表明,3岁以下的儿童被正式诊断为自闭症是极其罕见的。然而,其他研究表明,可靠的诊断可以在2-3岁和成年期之间进行。
鉴于在诊断模式和诊断手册的不一致性方面存在相对不同的共识,对遗传和神经生物学前因和特性的研究激增,这将提供一个更强大和可复制的筛查。因此,许多研究声称,这种疾病具有强烈的遗传联系,以及已知的大脑变化,从出生起就明显存在。
这篇综述旨在将使用新技术的研究和历史研究结果统一到“常见的神经生物学机制”中,这可能有助于诊断和新的治疗。方法
研究人员回顾了与ASD有关的生物学机制的现有数据。作者将重点放在“已经影响临床实践”或“可能在未来10年内影响临床实践”的问题上
这篇综述分为几个部分,分别讨论了风险(遗传联系和候选资格、环境因素)、“大脑发育改变”、“自闭症谱系障碍模型系统”,以及“未来方向”。总的来说,它参考了来自各种期刊的113种不同的证据。 结果
研究基础表明,ASD具有较高的遗传成分,同卵双胞胎的一致性约为50%,非同卵双胞胎的一致性高达30%。据报道,兄弟姐妹之间的总体发病率估计为25%,如果有两个或两个以上的兄弟姐妹患有自闭症,或者其中一个受影响的是女性,那么发病率将上升到50%。常见的遗传变异具有一定的解释力,但对风险的总体贡献很小,这表明风险随着更多基因的存在而增加。ASD与罕见遗传综合征的关联研究提供了一些明确的风险生物学标志物。到目前为止,基因组广泛关联研究的使用还没有显示出比特定变异的风险更大的结果。因此,利用微阵列、测序和基因组分析已经发现了denovo微缺失和微复制的证据,这暗示了可能的致病潜力。这些是遗传模式的微小变化。这其中包括12种拷贝数变异(表2),以Cr15、16和22上的异常为主。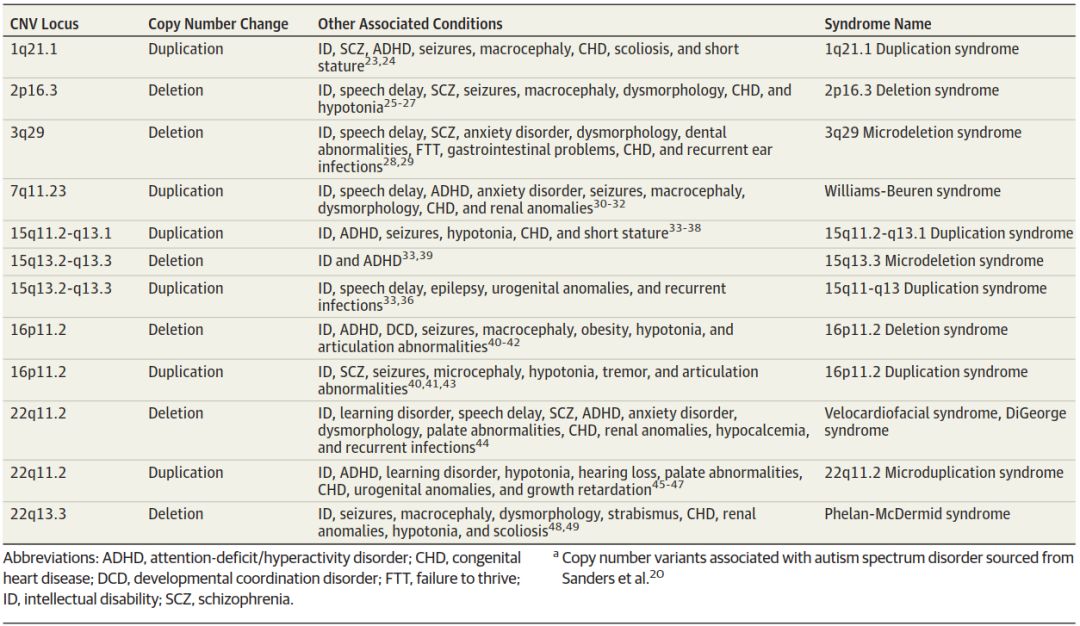
本综述还将更多新的研究技术引入基因(外显子组)的蛋白质编码区域,阐明了单核苷酸变异和插入或缺失的异常,显示ASD患者中异常的比例更高。因此,他们指定了24个细胞遗传学位置,通过从离子转运异常到DNA修复的代谢破坏方法,增加了ASD发生的风险(表3)。与测序结果不同,这些外显体差异的染色体位置更具全局性,提示ASD的发生机制复杂多样。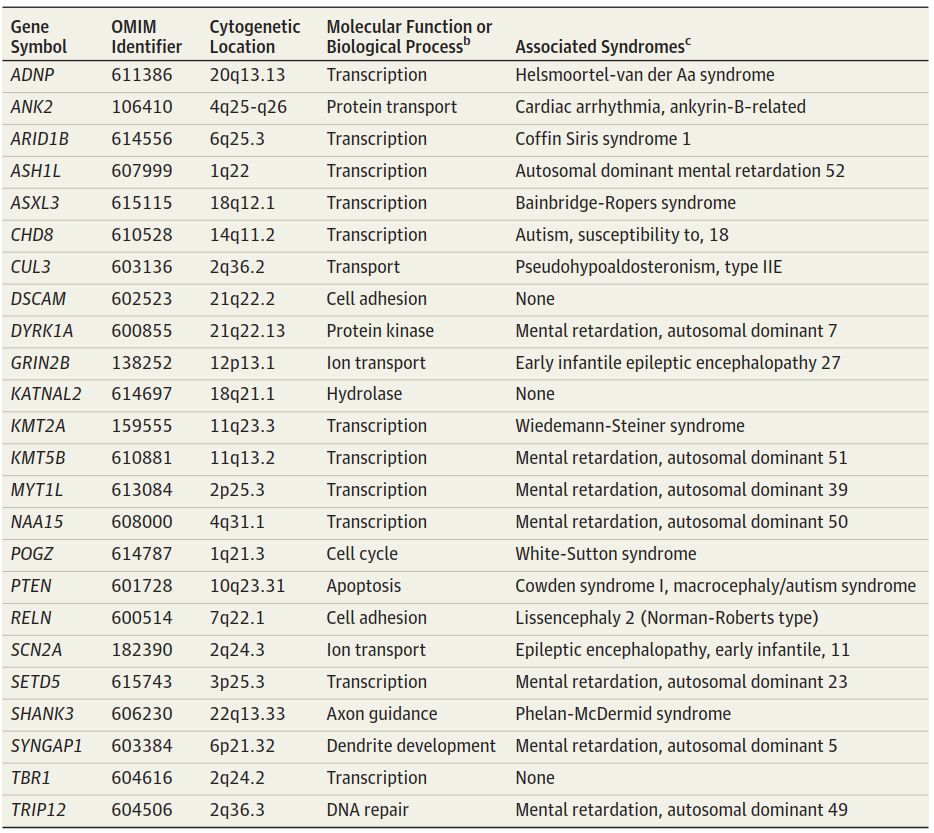
作者将这些基因分成两大类,一类涉及大脑功能,另一类涉及基因调控。此外,计算模型已经发现,这些基因中的一些共同表达与早孕至中期特定脑区的ASD有关。总之,作者推测ASD的遗传风险可能是由于多个基因通过不同的机制相互作用。婴儿时期增加ASD终生风险的因素包括母体条件、妊娠并发症、药物和暴露于有毒物质(不包括许多人声称的疫苗),在这一关键时期,暴露有毒物质会影响神经发育。尽管如作者所指出的那样,由于各种原因,难以将风险分配给环境,但也有一些很强的风险因素。在每10年的年龄增长中,提高父母年龄是ASD的一个独立危险因素(母亲占18%,父亲占21%)。这可以通过引用父母提供的基因的新生突变来解释,这种突变随着年龄的增长而增加。早产也是一个风险因素。可能导致潜在脑细胞损伤的具体机制包括与早产有关的机制(缺氧、低血糖),这在急性神经发育阶段(例如神经元迁移)中可能更为重要。进一步的风险可以细分为“内源性”(短期间隔妊娠、分娩并发症,包括低氧血症、缺血或创伤、孕妇肥胖和糖尿病)或“外源性”(药物暴露,如丙戊酸钠、b2肾上腺素能哮喘药物,以及潜在的空气污染)。作者的结论是,促进ASD风险的损伤机制可能直接作用于脑细胞和支持细胞,也可能通过炎症或激素干扰间接作用。他们认为环境因素实际上可能影响基因,这表明不同的危险因素之间存在复杂的相互作用。评估脑发育改变的主要方法是影像学,它可以包括非特异性的测量,如计算机地形图(CT)或更详细的分析,如功能磁共振成像和灌注加权成像(后者对描述精神疾病或其他认知过程的差异是很重要的)。作者在大脑发育异常的背景下提出了目前的研究结果,表明大脑细胞连接异常在ASD患者及其兄弟姐妹中普遍存在。持续的发现是大脑发育轨迹的变化,而不仅仅是神经结构的特定变化。这些数据表明突触生长和神经元修剪之间的不平衡可能参与其中。最近的神经影像学显示,早在6个月大的时候,参与感觉处理的神经连接和神经回路的发育变化可能就被破坏,一些研究表明,这种破坏的严重程度与未来症状的严重程度之间存在联系。目前一致的发现包括,从6 - 12个月开始,灰质区域(尤其是与听觉和视觉处理相关的区域)扩大,然后在12-24个月之间出现普遍的“过度生长”。这种发育以一种“夸张”的方式,并不代表有益的神经元发育,或者可能代表了大量未经过修剪的残余神经元。随着年龄的增长,ASD患者的总体脑容量继续超过他们的同龄人,尤其是杏仁核、额叶皮质和颞叶皮质内与社交、情感、情感处理、显著性和语言相关的区域。到了14岁,ASD患者和非ASD患者的大脑相对大小的差异就消失了,但很明显,这种快速的扩张并没有导致同样的结果功能。事实上,核磁共振成像已经表明,在整个学校教育、青春期和成年期都存在着持续的中断,与对照组相比,这种异常的连接模式不仅与连接有关,而且与大脑各区域之间有序和整合连接的过程有关。作者进一步证明,16p11.2缺失综合征患者在发育和宏观结构上表现出类似的变化,并将其与ASD的关系作为这两种情况的病理学证据。作者还回顾了计算(即预测)细胞和动物模型的研究结果,这些模型可以提供关于个体和高度特异性过程的信息,也可以提供串联过程一般方向的信息。他们指出,斑马鱼与人类有24种ASD候选基因相同,这对研究神经发育过程非常有用。对大鼠和灵长类动物的研究也显示,它们可能表现出类似于自闭症患者的行为,例如,在基因表达与Rett综合症类似的猴子中,社交接触减少。对共享16p11.2基因重叠的小鼠行为的研究表明,小鼠的某些行为与人类ASD症状一致,包括多动、重复行为和学习困难。小鼠模型也显示出长期增强能力减弱(一种理论上的学习机制),这显示出对药物的一些反应。用分离的人类细胞系进行的实验也显示出了希望,16p11.2中基因的显著表达显示出可预测的过剩和匮乏,依赖于细胞系内外CNVs的复制或缺失(偶然的负变异)。对成熟为神经元的多能干细胞的进一步研究正在进行中。作者继续围绕诊断和早期治疗提出了一些建议,主要关注前者的观察标准和对后者的支持。他们推测,目前遗传关联的证据为治疗提供了可能的途径,同时也提供了一种分子诊断的形式,但他们承认,目前针对基因位点的特定治疗可能会产生无法预测的结果,导致除了最严重的病例外,所有病例的治疗都无法进行。
在诊断方面,他们指出,个别的发现,无论是神经生物学的还是遗传学的,都不太可能得到证实,但如果存在多种变化,则可能意味着风险增加。他们评论说,尽管目前诊断ASD的方法确实汇集了共同的核心症状,但由于症状和原因的个体差异,在这方面存在一些方法上的限制。
本研究显示,ASD可能不是一种单一的疾病,而是行为症状和原因的复杂相互作用,精确医学和研究可能在未来提供治疗。作者建议,在等待新研究和新技术的同时,我们把重点放在早期诊断和行为治疗上,药物只用于伴发症状。他们还建议筛查常见的相关遗传疾病,如脆性X染色体。除此之外,还有更多的一些临床建议:
咨询自闭症患者,包括对“异常”或“正常”的现实判断。
考虑对兄弟姐妹中的高风险候选基因进行基因检测,可能有助于早期诊断和管理。
对异常神经元发育的研究方向可以促进新技术的发展,不仅限于研究ASD,还可以研究其他神经发育、遗传、精神和神经疾病。
关注行为方面的治疗,而不是基于诊断的预期治疗结果。
- 基因编辑的作用虽然令人兴奋,但还需要进一步考虑意外结果。这是一个非常微妙的领域,任何治疗都需要来自个人、倡导团体的大力支持,并需要严格的立法。
- 功能神经成像方法的改进,包括利用可追溯的同位素绘制神经元迁移、增殖和修剪的潜在用途。这应该仅限于体外研究,并且只有在征得同意并经过与伦理小组的商议讨论后,才可以考虑在人类身上进行。在每一种情况下,使用任何同位素都不应造成伤害,并且只能对指标情况和整个群体产生总体效益。
- 对妊娠期风险较高的人群进行咨询,以帮助早期诊断和后续支持。
参考文献
Muhle RA, Reed HE, Stratigos KA,Veenstra-VanderWeele J. (2018) The Emerging Clinical Neuroscience of AutismSpectrum Disorder: A Review. JAMA Psychiatry. 2018;75(5):514–523.doi:10.1001/jamapsychiatry.2017.4685